pysimm integrates with two well-renowned tools for structural and topological analysis of molecular systems.
Either you prefer to use Zeo++ from Dr. M.Haranczyk or you used to work with Poreblaser
developed in Dr. L.Sarkisov's group in both cases you can setup structural analysis of your samples in pysimm
in a consistent and fully automated way.
Python Simulation Interface for Molecular Modeling
Recent News:
Release v1.1: CHARMM support and polymers tacticity
|
PySIMM version 1.0 release
|
Zeo++ interface in new version 0.2.3


About
![]() pysimm is an open-source object-oriented Python package for molecular simulations.
It handles data organization for particles, force field parameters, and simulation settings
so you can focus on developing your simulation workflow. For a detailed reference document, see the
Documentation page.
pysimm is an open-source object-oriented Python package for molecular simulations.
It handles data organization for particles, force field parameters, and simulation settings
so you can focus on developing your simulation workflow. For a detailed reference document, see the
Documentation page.
Integration with dedicated software for geometry analysis
Force Field Assisted Linear Self-Avoiding Random Walk
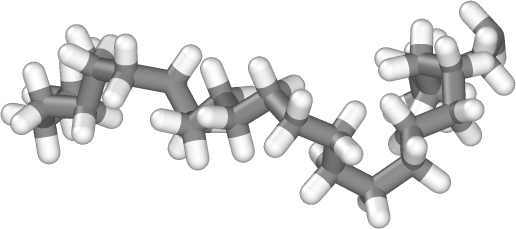 Create long linear polymer chains structures modeled with common atomistic force fields.
The random walk application in pysimm makes it easy to turn a reference repeat unit into a system of long, linear polymer chains.
See the random_walk application
section of the reference document for more information.
Create long linear polymer chains structures modeled with common atomistic force fields.
The random walk application in pysimm makes it easy to turn a reference repeat unit into a system of long, linear polymer chains.
See the random_walk application
section of the reference document for more information.
LAMMPS Integration
![]() pysimm integrates smoothly with LAMMPS for performing large-scale particle based molecular
simulations. Although still fully customizable, many simulation parameters are intelligently
inferred from a molecular system definition to simplify the simulation setup process. You can choose to
prepare raw LAMMPS input files yourself or use pysimm classes to easily set up complex simulation algorithms.
pysimm integrates smoothly with LAMMPS for performing large-scale particle based molecular
simulations. Although still fully customizable, many simulation parameters are intelligently
inferred from a molecular system definition to simplify the simulation setup process. You can choose to
prepare raw LAMMPS input files yourself or use pysimm classes to easily set up complex simulation algorithms.
The interaction with software packages uses an abstract object-oriented design that allows simulations to be provided as adjustable parameters for applications. Simulation Python objects can be swapped in and out of application workflows. See the lmps module section of the reference document for more information.
Iterative Monte Carlo-Molecular Dynamics workflows
The abstract Simulation architecture has been extended to support simulation with the Cassandra and RASPA monte carlo
software packages. Grand canonical monte carlo simulations can be performed on pysimm System objects, enabling iterative
Monte Carlo-Molecular Dynamics workflows to study the relaxation of materials upon adsorption of small molecules.
See the examples in the source code repository and the documentation for more information.
Support for Common Atomistic Force Fields
![]() pysimm includes parameters for various atomistic force fields in an organized, searchable database that can be
programatically used to update topology information for pysimm system structures. Choose to use simple typing rules
to assign particle types, or manually choose the appropriate ones. Then let pysimm do the rest of the work, automatically
updating bond topologies from the force field database. See
the forcefield package section of
the reference document for more information.
pysimm includes parameters for various atomistic force fields in an organized, searchable database that can be
programatically used to update topology information for pysimm system structures. Choose to use simple typing rules
to assign particle types, or manually choose the appropriate ones. Then let pysimm do the rest of the work, automatically
updating bond topologies from the force field database. See
the forcefield package section of
the reference document for more information.
Work with pyIAST
pyIAST is an open-source Python package which provides implementation of the Ideal Adsorption Solution Theory. It can fit simulation or experimental adsorption data into analytical isotherm models. Is easy to integrate and use within pysimm framework for visualisation, interpolation, and characterization of simulated pure-component adsorption isotherms.
Open Source Development
The source code and development can be viewed at our GitHub repository. pysimm is licensed under the MIT License.
If you use pysimm, please cite:
- Fortunato, M. E.; Colina, C. M. pysimm, https://github.com/polysimtools/pysimm.
- Fortunato, M. E.; Colina, C. M. “pysimm: A Python Package for Simulation of Molecular Systems.” SoftwareX, 2017, 6, 7-12. [doi]